SeqCrawler provides a DBI interface as a Perl module to link GBrowse2 with the Solr index.
A simple configuration in gbrowse specific configuration file is needed to do so:
db_args = -adaptor solr
-STORAGEURL http://my_riak_external_hostname:riak_port/riak
-BANK GenBank (optional, default is all)
Available options are the following
db_args = -adaptor solr
// Following is optional
-port (default: 8080, solr server port)
-host (default: localhost, solr server host)
-maxresult (default: 99999999, max mnuber of returned results)
-BANK (default: all, bank name in index as a filter)
-STORAGEURL (default: undefined, url to the sequence backend. If not set, dna or protein sequences will not be retrieved)
The system is preconfigured for a number of features (rna, cds, ...). However, if indexed data adds new features, the gbrowse configuration should be updated to specify their display. To do so, edit the seqcrawler configuration file (/etc/gbrowse2/gbrowse.conf/seqcrawler.conf) and append the new features (see GBrowse configuration manual). Example:
[chromosome] feature = chromosome glyph = segments bgcolor = lightslategray fgcolor = black key = chromosome citation = chromosome category = Analysis das category = experimental
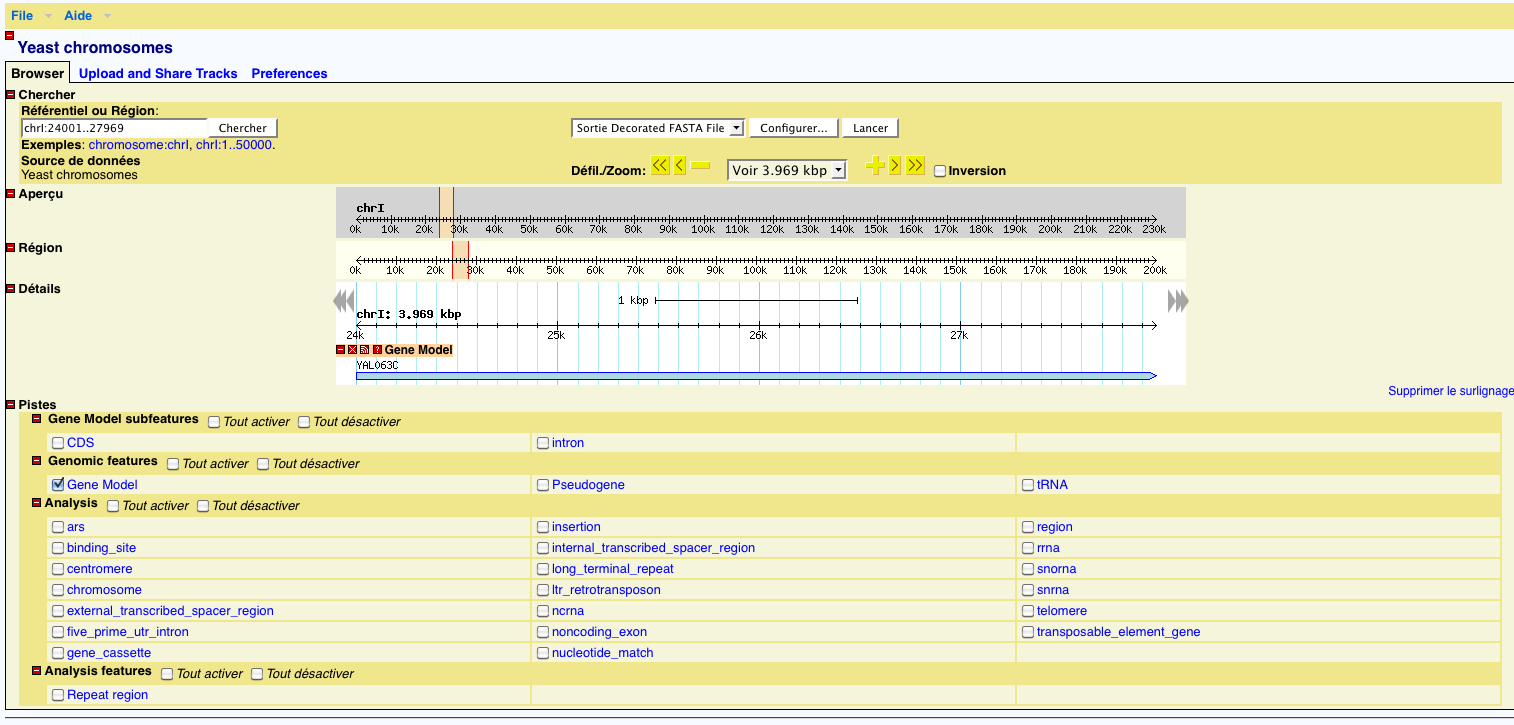
GBrowse provides a web interface to visualize a genome and browse it. It is interfaced to the index to get GFF features for a chromosome with positions etc... The user can click on a gene to gets its details, or change the select area. There are also some export functions available.
The interface and behavior is customizable help with a very functional configuration file. For more information, refer to the GBrowse tutorial.