
This image hosts the following software:
Those softwares are pre-configured and ready to run or to add new data to index.
All required packages are preinstalled.
The goal here is not to get an administration guide of solr or gbrowse with all there configuration stuff, but only to server as a guide on how to use it in this image as well as some tips to use it. For further information on each software configuration, please refer to their respective documentation.
Solr is an indexing platform based on Lucene. It provides API to index/query as well as a basic web interface. The Solr query syntax is available at http://wiki.apache.org/solr/SolrQuerySyntax Queries can be made against whole indexed text or against specific fields (id, accession... fields depends on indexed documents). Query ranges are also supported for a few parameters (start and end for gff documents for example). Solr can be queried vie basic HTTP requests and can return results in Xml or JSON format. Solr is installed under /opt but also as a web application in Tomcat.
Riak is a NOSQL backend used to store raw data. It also provides a web interface to query data based on their id. Riak is a cluster of nodes acting as a ring with data replication to provide data high availability. To start or stop riak use:
riak start/stop
Riak is an other NOSQL backend used to store raw data. It also provides a web interface to query data based on their id. MongoDB is a cluster of nodes acting as a ring with data replication to provide data high availability. To start or stop MongoDB use:
service mongodb start/stop
Add repository osallou.genouest.org/apt
Run:
apt-get update apt-get install seqcrawler
Dependencies (mongo, gbrowse) will be automatically installed.
Package install all config files in /etc/seqcrawler (both for GBrowse and Seqcrawler itself).
Web servers are available at:
http://myhostname:8080/seqcrawler/
http://myhostname/mongo/
http://myhostname/gb2/gbrowse
Indexes: /var/lib/seqcrawler/index
Access to admin section (/admin) should be restricted to a specific role, defined in tomcat user configuration.
Access to port 8080 and 8098 (respectivelly Tomcat and Riak, if used) should not be allowed to unauthenticated users, as HTTP PUT and DELETE operations can be used to operate on index and raw data backend. Users should access via the port 80 with a web proxy defined in Apache configuration towards those ports.
A default index is available with the Yeast genome in seqcrawl-example package. The data comes from Genbank.
To index some data, a schema file describing the index must be updated first
For indexing usage:
bin/crawler.sh -h
Main options are:
-t : to define the type of data to index according to known types (Fasta, Embl, GFF, Readseq for others)
-b: bank name (defaults to GenOuest).
-C: to clear the index before inserting new data
-c: to clear data for input bank
TIPS:
Indexing large data can be time consuming. In such a case, it is advised to index small parts of data then, and only then, to merge the indexes.
It is not possible to run at the same time multiple index operations on the same index directory.
To merge some indexes, use the script merge.sh. For usage:
bin/merge.sh -h
Using GBrowse makes sense of course only if data represent position on a sequence, and that several data elements are positioned on the same sequence.
To use both tools, data must be in GFF3 format. Some tools and APIs are available to convert to this format (BioJava,BioPerl, readseq).
A Genbank to GFF conversion tool is available on the image, else readseq is installed on the image.
To use the Genbank converter, data must be in gzip format (.gz):
cd /usr/share/seqcrawler/solr/apache-solr/seqcrawler/solr/bin/ perl genbank2gff3.pl -d mydirectorywithGenbankfiles -o mygffoutputdir
To index GFF files and make them available both via indexer and gbrowse, the data must be indexed as usual and a Gbrowse configuration file must be created (no order in creation)
It is possible to have a single configuration file for all banks, or one per bank.
A sample configuration file is available at /etc/gbrowse2/seqcrawler.conf.
Customization concerns:
db_args = -adaptor solr
-FORMAT gff
-STORAGEURL -STORAGEURL http://localhost/cgi-bin/mongo/mongo.pl?id=
# -BANK GenBank
STORAGEURL parameter should be updated to local host (external interface name)
The BANK parameter is optional, and can be used to limit displays to a single bank.
For Gbrowse customization (header footer etc...), please refer to the Gbrowse documention at Gmod.
Once created, Gbrowse is accessible via URL:
http://hostname/gb2/gbrowse/nameofmyconffile/
After the addition of a new index, or the modification of an index property file, the new data must be (re)loaded.
To do so, either restart tomcat or reload web application in Tomcat Manager.
This is main Gbrowse configuration file.
[General section] specifies the path to main directories used by the software.
Render_farm parameter is activated by default in this configuration to enhance performances.
Custom HTML elements can be set here according to desired look of the browser. CSS elements are in the web directory.
User account support is not activated.
To manually add a new bank, a specific bank section must be added like:
[mybank] description = my test bank path = /etc/gbrowse/gbrowse.conf/mybank.conf
All features are not represented in provided configuration file. If some features are missing based on new indexing data, those can be added at any time like the following:
[transposable_element_gene] feature = transposable_element_gene glyph = segments bgcolor = lightslategrayfgcolor = black key = transposable_element_gene citation = transposable_element_gene category = Analysis das category = experimental
Simply update title, feature, key, citation to have a new graphical representation of the feature. Glyph and gbcolor can be updated to change the color and way of display. Refer to other examples and Gbrowse documentation for additional details.
Before going in configuration details, here is a short introduction on how on index is created.
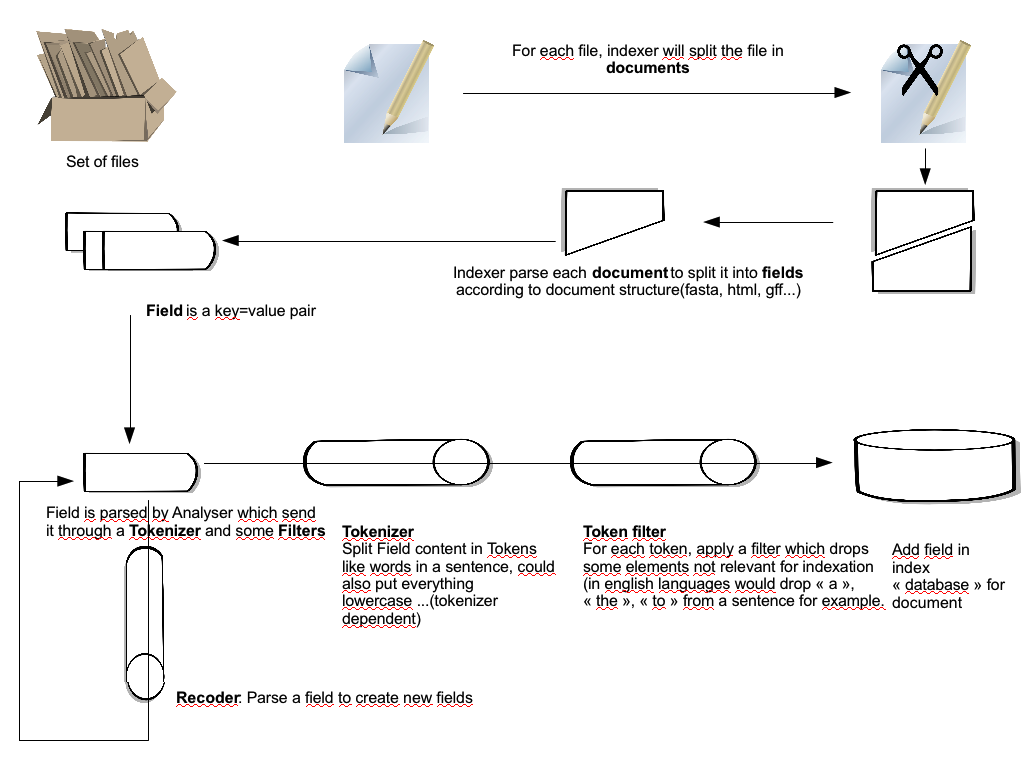
To index some files a schema configuration file is used..
Configuration files are located in /etc/seqcrawler/solr
The global property file is solrconfig.xml. It holds configuration for the indexation platform such as index root directory. Many properties are available, one gonna describe only major configuration data required for directories or server cusotmization. For other properties (may impact performances), please refer to solr documentation.
Main properties:
<dataDir>${solr.data.dir:/index/data}</dataDir>
<requestHandler name="shard" class="solr.SearchHandler">
<!-- default values for query parameters -->
<lst name="defaults">
<str name="echoParams">explicit</str>
<!-- <str name="shards">shardserver1/solr/,shardserver2/solr/,...</str> -->
</lst>^M
</requestHandler>
In specified request handlers, uncomment the shards field is index shards is required. Specify the hostname or ip address of the servers handling the shards separated by a comma.
Facet fields provide an additional result field with the count of terms for the query.âfacet.fieldâ specify which field index to facet. There can be multiple facet.field, but this method is RAM consuming depending on index size and terms count. In this example, for the total of matches, it will count the bank field terms and the organism field terms. Facteted fields are automatically written in Solritas web interface with given templates.
The schema file must be the same for all the shards. They can differ for different indexes. It specifies how to index and analyse the data in the index.
FieldTypes are predefined Types with Tokenizer etc... It is possible to create new FieldTypes based on existing ones or with new Java classes.
Then fields are defined based on those FieldTypes. Many examples are provided in the schema to help at customization.
Depending on new indexes, additional values may be required in this schema if default dehaviour (see dynamic field) is not appropriate.
#Fields for GFF indexing: <field name="bank" type="lowercase" indexed="true" stored="true" default="genouest"/> <field name="seqid" type="uuid" indexed="true" stored="true" default="NEW"/> <field name="id" type="lowercase" indexed="true" stored="true"/> <field name="chr" type="lowercase" indexed="true" stored="true"/> <field name="feature" type="lowercase" indexed="true" stored="true"/> <field name="start" type="int" indexed="true" stored="true" multiValued="true"/> <field name="end" type="int" indexed="true" stored="true" multiValued="true"/> <field name="strand" type="lowercase" indexed="true" stored="true"/> â¦. <field name="stream_name" type="lowercase" indexed="false" stored="true"/> <field name="stream_size" type="lowercase" indexed="false" stored="true"/> <field name="stream_content_type" type="lowercase" indexed="false" stored="true"/> ⦠<field name="text" type="text" indexed="true" stored="false" multiValued="true"/> ⦠<dynamicField name="*" type="text" indexed="true" stored="false"/> ⦠<copyField source="*" dest="text"/>
It is possible to search an index with a query without the web interface. To do so, the java class org.irisa.genouest.seqcrawler.query.Query is available. See usage:
java -classpath $INDEXHOME/CrawlerIndex-X.Y-jar-with-dependencies.jar org.irisa.genouest.seqcrawler.query.Query -h
This very simple query interface will log to console the first results of query.
To search via a browser, refer to Solr query syntax
Access to http//host/cgi-bin/mongo/mongo.pl with parameters:
source: orignal source raw data (e.g. nucleic sequence chromosome for example hosting a gene)
id: additional raw data for the element id (transcript for example).
start,stop: optional start and stop position of the sequence to extract in source
